
Синдром Жильбера (СЖ) – это генетически обусловленное заболевание печени, которое характеризуется повышением уровня билирубина в крови вследствие недостаточности фермента уридинглюкуронилтрансферазы 1А1 (УГТ 1А1). Впервые данный синдром был открыт во Франции в 1901 году гастроэнтерологом Огюстеном Николя Жильбером. На протяжении нескольких десятилетий изучались исходы заболевания и его влияние на здоровье в целом. В наше время интерес к СЖ возрождается, так как появилась возможность объективного генетического подтверждения диагноза. Данный синдром представляет интерес большому кругу врачей. Как педиатры, так и гастроэнтерологи-гепатологи довольно часто встречаются с этим заболеванием в своей практике.
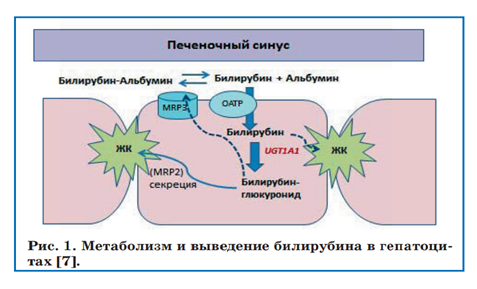
В основе СЖ лежит нарушение конъюгации билирубина (рис1). После разрушения эритроцитов и гемсодержащих белков образуется несвязанный билирубин (НБ), который связывается связывается с альбумином. Таком комплекс циркулирует в плазме, затем в печени НБ связывается уже с лигандами и проникает в цитозоль с помощью белков-переносчиков. В эндоплазматическом ретикулуме происходит конъюгация билирубина в моно- и диглюкуронид с помощью фермента глюкуронилтрансферазы (UGT-1). Образуется связанный (прямой) билирубин, который выделяется в желчный каналец с помощью транспортных белков. [1]
Раннее, до генетической идентификации СЖ, диагноз выставлялся на основе клинических признаков и лабораторных исследований. К клиническим проявлениям относятся боли в животе, расстройство аппетита, субиктеричность кожи и иктеричность склер. Данные семейного анамнеза часто говорят о наследственной предрасположенности. Из лабораторных данных обнаруживается изолированное повышение уровня билирубина в крови до умеренных значений, в пределах 21–85 мкмоль/л, без нарушения других биохимических показателей функции печени. Наличие признаков гемолитической анемии и маркеров гепатитов при повторных исследованиях отсутствует.
Современное подтверждение диагноза СЖ основывается на генетическом исследовании. Генетический дефект СЖ появляется из-за дополнительного динуклеотида ТА на промоторном участке А(ТА)6ТАА гена, кодирующего фермент уридилдифосфатглюкуронилтрансфераза (УДФГТ). Это, в свою очередь, ведет к образованию участка А(ТА)7ТАА. Удлинение промоторной последовательности ведет к снижению образования УДФГТ. Эта мутация обозначается UGT1A1*28 и является наиболее распространенной и изученной. В современной практике генетиков имеются данные о наличии до 113 вариантов мутаций данного гена (UGT1A1*1-*113).
Для пациентов с диагностированным синдромом Жильбера особое внимание следует уделять гепатобилиарному тракту. Известно, что СЖ играет определенную роль в формировании желчнокаменной болезни (ЖКБ) в популяции. Известно, что ЖКБ страдают преимущественно женщины, но наличие СЖ делает уязвимыми и лиц мужского пола. На сегодняшний день можно считать доказанным, что обладатели СЖ — контингент риска развития ЖКБ.
Также следует отметить, что некоторые токсические метаболиты лекарственных препаратов могут провоцировать развитие гипербилирубинемии с внутрипеченочным холестазом. Это происходит из-за повреждения повреждения белков-переносчиков, экспортирующих желчные кислоты, либо белков-транспортеров, участвующих в экспорте билирубина и других молекул из гепатоцита в желчные канальцы. К таким препаратам относятся андрогены и эстрогены, цитостатики, хлорпромазин, сульфаниламиды, полусинтетические и синтетические пенициллины, макролиды, цефалоспорины, блокаторы гистаминовых рецепторов, пероральные сахароснижающие препараты – производные сульфанилмочевины и прочие.
Для превентивного лечения неблагоприятных последствий синдрома Жильбера и лечения холестаза, задачей терапевтического воздействия является снижение уровня билирубина — как непрямого (свободного), так и прямого (связанного). Снижению непрямого билирубина способствует фенобарбитал, повышающий активность ГТФ, назначаемый в возраст - ных дозах непосредственно или в составе Валокордина. Для снижения уровня прямого (связанного) билирубина целесообразно применение урсодезоксихолевая кислота (Урсофальк, Урсосан). УДХК стимулирует как канальцевые белки-транспортеры, так и базолатеральные транспортные белки, что индуцирует выведение токсических субстратов из гепатоцитов, и оказывает защитное действие при поражении нервных клеток непрямым билирубином.
Список литературы
- Ипатова М.Г., Шумилов Петр Валентинович, Шагалова Д.Л., Нестерова Та., Иванова А.С. Особенности фармакотерапии у пациентов с синдромом Жильбера // Педиатрия. Журнал им. Г. Н. Сперанского. 2015. №6. URL: .
- Шомина К.П., Кряжева В.Р. Диагностика синдрома Жильбера на современном этапе // FORCIPE. 2019. №Приложение. URL: https://cyberleninka.ru/article/n/diagnostika-sindroma-zhilbera-na-sovremennom-etape.
- Farago B. Gilbert’s syndrome / B. Farago, B. Melegh // Orv. Hetil. 2008. Vol. 149. № 27. P. 1277–1282.
- Genetic variation in UGT1A1 typical of Gilbert syndrome is associated with unconjugated hyperbilirubinemia in patients receiving tocilizumab / J. S. Lee [et al.] // Pharmacogenet. Genomics. 2011. Vol. 21. № 7. P. 365–374.
- Рейзис А.Р., Хохлова О.Н., Никитина Т.С. Синдром Жильбера: современные воззрения, исходы и терапия. Доктор.ру. 2012; 3 (71): 42–45.



